分子动力学模拟是分子模拟中最接近实验条件的模拟方法,能够从原子层面给出体系的微观演变过程,直观的展示实验现象发生的机理与规律,促使我们的研究向着更高效、更经济、更有预见性的方向发展。分子动力学可以解决和研究DNA的折叠和性质、蛋白与配体的识别机制、跨膜蛋白的工作机理、蛋白酶与底物的反应、蛋白与蛋白的耦合、比较野生型与突变蛋白的不同特性、蛋白折叠的机制问题(控制温度,使蛋白自行折叠和去折叠)等。
本研讨班旨在研究对象模型的获取与构建-体系预处理、能量优化、分子动力学模拟结果评估、结合自由能计算、相互作用机理分析、可视化、轨迹特征获取,并对经典文献进行复现。
本次研讨班分为2个阶段
1. 分子动力学模拟的研究思路
以实际案例和高分论文为例,讲解分子动力学模拟研究思路
2. 掌握分子动力学模拟流程
以实际结构为例,详细介绍分子动力学的原理、软件过程及后续结果分析。
主办单位:北京市计算中心有限公司
协办单位:
北京市基因测序与功能分析工程技术研究中心
云计算关键技术与应用北京市重点实验室
工业和信息化人才培养工程培训基地
北京市大数据教学实践基地
举 办 地:北京市海淀区丰贤中路7号北科产业3号楼
课程安排:2024年2月29日-3月1日(周四-周五) 上9:30-11:30 下13:30-17:00
|
日期 |
主题 |
内容 |
备注 |
|
第一天 |
分子动力学模拟简介 |
1. 分子模拟的发展历史 2. 分子模拟的优势和局限 3. 分子动力学基本原理 |
理论+ 操作 |
|
Linux基础 |
1. Linux服务器连接 2. 数据传输 3. Linux常用命令 |
||
|
动力学模拟预备知识 |
1. Amber软件介绍 2. Amber力场介绍 3. 拓扑文件、坐标文件介绍 4. 模拟条件介绍 1) 系综(ensemble)选择 2) 约束条件设置 3) 参数配置 |
||
|
研究模型预处理 |
1. 模型文件的预处理 2. 结构文件来源介绍 3. tleap模块的介绍和使用 1) 蛋白预处理 2) 小分子预处理 3) 溶剂化模型的构建方法 4) 生成amber识别的prmtop和inpcrd输入文件 |
||
|
第二天 |
分子动力学模拟流程 |
1. 动力学模拟参数文件的介绍 2. 能量优化 3. 升温 4. 平衡 5. 动力学成品模拟 |
理论+ 操作 |
|
分子动力模拟结果分析 |
1. Amber轨迹文件处理 1) 轨迹合并 2) 采样、重设时间、截取部分原子、删除水和离子、成像居中和构象叠合 2. 结合自由能计算 3. Amber轨迹分析 1) RMSD分析 2) RMSF分析 3) B因子分析 4) 氢键分析 5) 回转半径分析 |
理论+ 操作 |
|
|
生物分子结构可视化软件介绍与使用 |
1. VMD:结构可视化软件操作演练 2. VMD查看分子动力学模拟结果 3. VMD作图实例讲解与练习 |
理论+ 操作 |
注:内容以实际发生为准;若调,会提前通知。
示例:
动力学模拟流程介绍:
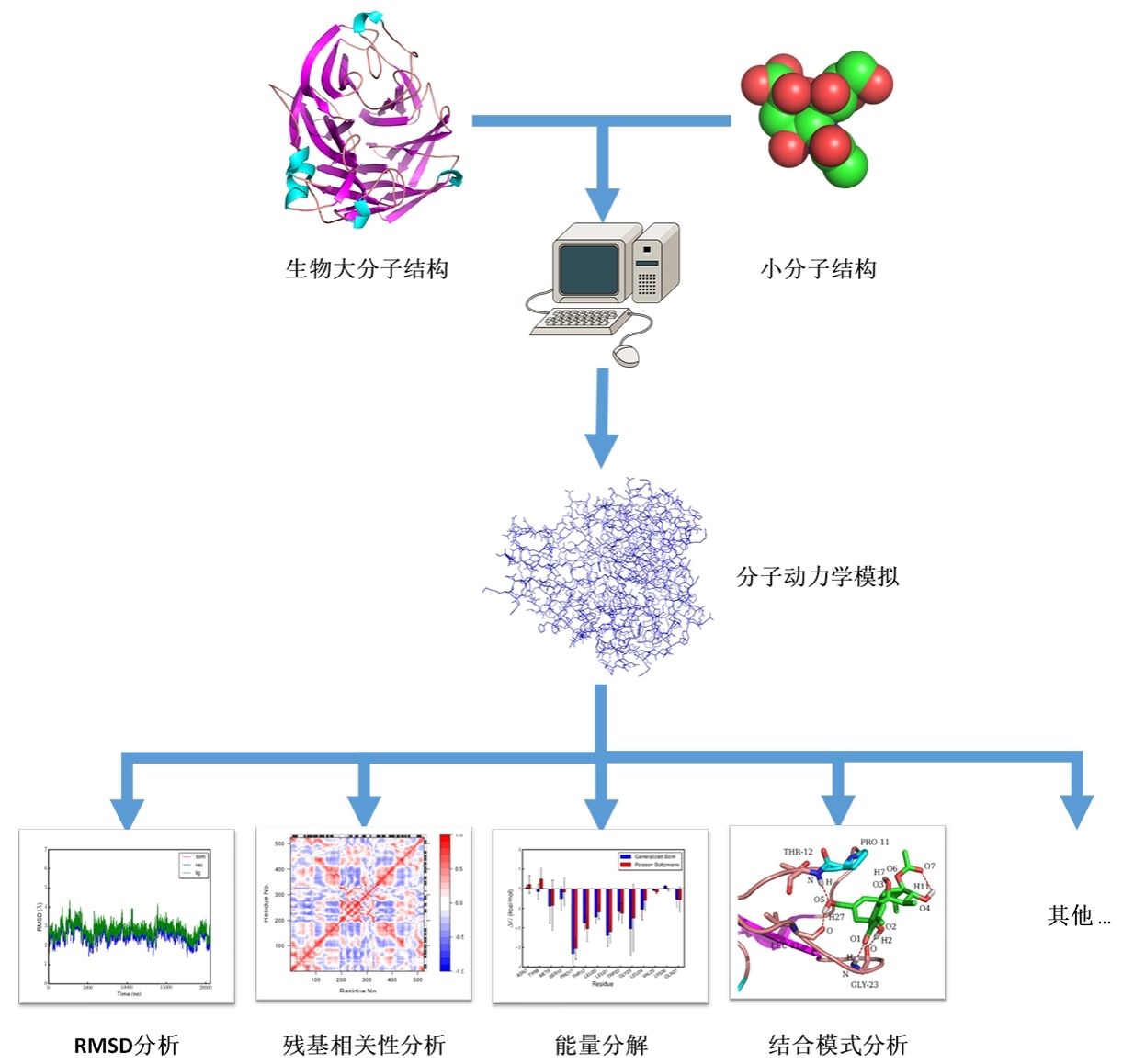
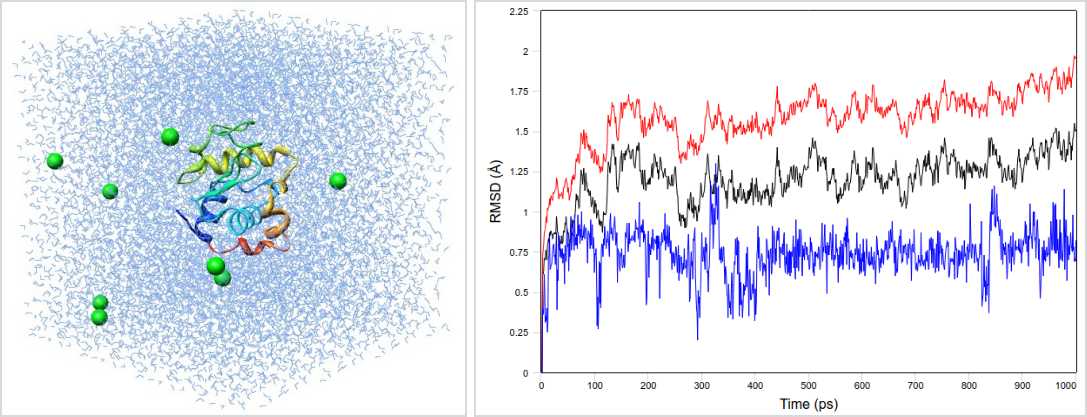
模拟体系结果展示:

【报名费用】
注册费:2800元/人(含当期听课费、资料费、证书费、考试费(如有))。
提供当期视频回放以供复习使用(羽林学院平台)。
开具增值税发票,提供盖章通知、结业证书等相关材料。
【报名优惠政策】
1、3人以上团体报名每人可减少300元;
2、4+1团报,可免费赠送一个名额;
3、上面优惠政策不能同时享受,只能享受其中一种;
老学员参加及推荐学员参加均可额外优惠200元。
【报名回执】
【咨询请联系】
QQ号:2814500767
徐老师 010-59341786,15801436028(微信同号)
员老师 010-59341773,18701529461(微信同号)
【注】开课前一周会发送邮件通知;若未接到邮件通知,请电话咨询。